Running OpenMMDL Analysis
This page details the variables required to run the analysis and showcases the application of OpenMMDL Analysis.
OpenMMDL Analysis can be used to analyze MD trajectories for receptor-ligand interactions.
Variables
OpenMMDL Analysis consists of mandatory and optional variables. The following are listed down below:
Mandatory:
-t = topology file of the simulation (in .pdb format)
-d = trajectory file of the simulation (in .dcd format)
Optional:
-n = Ligand name (3 letter code in PDB)
-l = Ligand in SDF format
-b = binding mode threshold. Is used to remove interactions under the defined procentual occurence from the binding mode generation. The default is 40% (accepted values: 0-100)
-df = Dataframe (use if the interactions were already calculated, default name would be "interactions_gathered.csv")
-f = final frame of the analysis (if you want to analyze only a certain part of the trajectory). The default will be the full simulation trajectory analysis.
-m = minimal transition threshold. Is used for the display of the binding mode transitions in the Markov state chains network figure. The default value is 1
-c = CPU count, specify how many CPUs should be used, default is half of the CPU count.
-p = Generate .pml files for pharmacophore visualization. The default is False (accepted values: True/False)
-s = special ligand name to calculate interactions with special ligands.
-nuc = Treat nucleic acids as receptor
-pep = Calculate interactions with peptides. Give the peptide chain name as input. Defaults to None
-ref = Add a reference PDB to renumber the residue numbers. Defaults to None (accepted values: str of PDB)
-r = Calculate the RMSD difference between frames. The default is False (accepted values: True/False) (if False no representative frame for the binding modes will be generated)
-w = stable-water-analysis. Defines if the analysis of stable water molecules should be performed. The default is False (accepted values: True/False)
--watereps = the EPS of the clustering part during the water analysis. will only result in something if "-w True" is added. Accepts float (in Angstrom).
--figure = File type for the figures, default is png. Can be changed to all file types supported by matplotlib.
--interaction_package = Interaction engine for protein–ligand contacts (choices: plip [default], prolif). Note: prolif is not supported together with -s (special ligands).
Application
An example of how a command line input for OpenMMDL Analysis should look like is:
If you need help with the command line input, you can always just use:
Results
The results of the analysis with OpenMMDL Analysis are stored in the current working directory.
You will obtain the following files and folders:
df_all.csv: This is the main source of raw data from the interaction analysis. It contains all the interactions that were found in the trajectory. Each row contains the information for one interaction of one frame.
- Barcodes:
This folder will contain figures of the barcodes for each interaction in the form of .png image files. Each interaction type will be stored in a separate file.

Furthermore the “Barcodes” folder will contain a subfolder called “Waterbridge_Piecharts”. This folder contains piecharts of the different waters interacting (with waterid) for each waterbridge interaction in form of .png image files.

- Binding_Modes_Markov_States:
This folder contains a .png image of all binding modes found in the markov state analysis aswell as .png images of the Markov chain plots.

This figure shows all binding modes found in the Markov state analysis. The binding modes are arranged by their occurence procentage in the trajectory. The 2D image of the ligand is coloured according to the interactions formed in this binding mode.
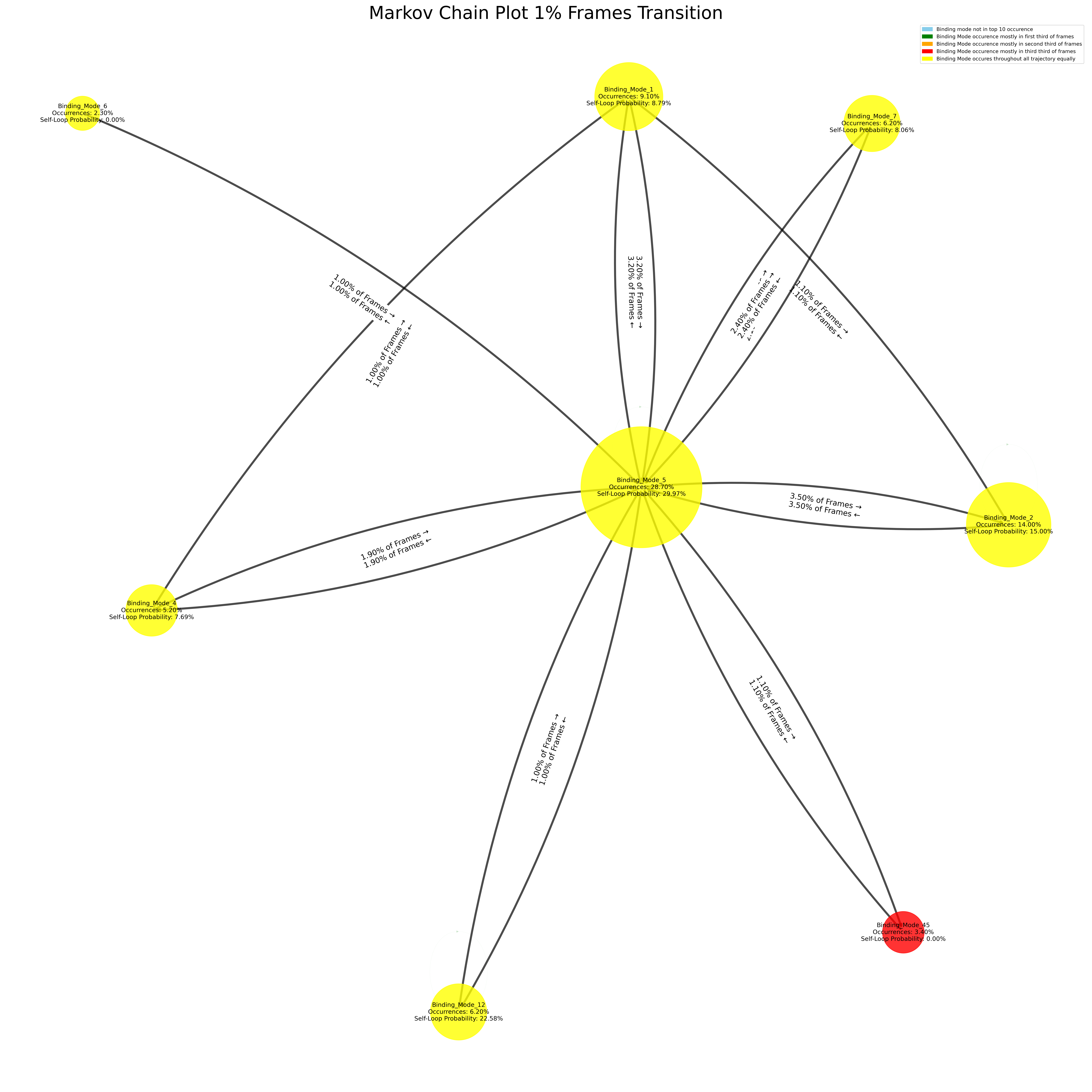
The Markov chain plot figures show the transition probabilities between the different binding modes. You will obtain four figures, each containing only transitions above a given cutoff. The cutoffs are 1%, 5%, and 10%.
- Visualization files:
These files are generated by OpenMMDL-Analysis for the visualization of interactions in form of point clouds. The files are:
clouds.json = contains the information for the point clouds
interacting_waters.pdb = topology file for the point clouds visualization
interacting_waters.dcd = trajectory file for the point clouds visualization
interacting_waters.pkl = pickle file of the interacting water ids for the point clouds visualization
Visualization
The interactions between your ligand and receptor can be visualized as interaction point clouds displayed ontop of your trajectory. Furthermore the visualization will display all waters that are involved in forming waterbridge interactions between your receptor and ligand. Open the visualization using the following command:
The command will open a prepared jupyter notebook in your browser. You will need to edit the following variables in the notebook (please note that the paths to the files need to be the absolute file paths):
After editing the variables, you can run the whole notebook and view the interactions in an NGL widget. Here is an example of the visualization

(CDK2 receptor with ligand LS3 (PDB: 1KE7))
Stable water analysis
This feature will analyze if within the MD stable water molecules are present. It will first collect all water molecules that move only slightly during the MD, then create clusters, where the cluster size is the EPS value given by –watereps (e.g. –waterepes 1.0, for clusters in the size of 1 Angstrom). All clusters are exported as PDBs with atoms at the position where a stable water molecule was present within the respective cluster. This will be performed for clusters present in 25% of the MD, 50%, 75%, 90% and 99% in separate folders. Furthermore, for each of these percentages one PDB with “representative water molecules” will be written. This contains one water molecule for each cluster. You can load this water PDB onto the protein. Lastly, the stable water analysis will output a csv file containing the interactions of protein residues with stable waters (using the representative water molecules). This function could potentially be called with any PDB file containing only water molecules and one PDB file containing a Protein (with or without ligand) and would result in a list of which residue might interact with which water molecule. Overall the stable water analysis might be useful for inhibitor optimization and determining structure activity relationship. Further information and example images are given within the OpenMMDL paper.